
Disulfide Bond Mapping Services
Talk to Our Scientists
"*" indicates required fields
Why do Disulfide Analysis?

Supporting data submission guidelines
Guidelines state: Sulfhydryl group(s) and disulfide bridges: If, based on the gene sequence for the desired product, cysteine residues are expected, the number and positions of any free sulfhydryl groups and/or disulfide bridges should be determined, to the extent possible. Peptide mapping (under reducing and non-reducing conditions), mass spectrometry, or other appropriate techniques may be useful for this evaluation.
Developability of protein
Understanding disulfide bonding is critical for protein characterization and in helping solve the structure. Incorrect disulfide bond formation or exchange can be side-effects of long-term mAb storage that can cause antibody aggregation. Providing early insight into the integrity of a biotherapeutic can allow identification of potential sources of error early on.
Scrambling will result in a misfolded protein which can lead to inactive, immunogenic or susceptible to aggregation.
Disulfide bonds can contribute to aggregation and must be monitored in regard to patient safety. In the biopharmaceutical production, elucidating the cysteine connectivities is necessary to prevent disulfide scrambling and folding incorrectly. Our services are typically used to identify the number and/or position of disulfide bridges present or to detect mismatched or scrambled disulfide bridge formation.
Mass spectra are compared before and after digestion to determine the location and type of disulfide bond present. DTT reduction is used to determine if the bond is inter-peptide or intra-peptide. Additionally, patterns within the spectra can be used to positively identify the presence of disulfide bonding.
Why Choose CovalX’s Disulfide Bond Mapping Service?
Experience
Founded in 2005, CovalX has over a decade of experience specifically focused on characterizing large molecules and protein interactions by mass spectrometry. Our primary focus is large biotherapeutic characterization, namely monoclonal antibodies and their derivatives.
Equipment
CovalX utilizes all the latest mass spectrometry equipment including Bruker, Thermo Orbitraps and Waters for both high-mass and high-resolution measurements. For front-end separation, we utilize several liquid chromatography platforms including both nano- and micro-LC separations. Additionally, CovalX utilizes the latest software packages from the mass spec manufacturers, third parties and custom-built software. Mass spectrometry is our specialty.
Trusted
CovalX is a trusted partner for nearly two decades with clients worldwide. We have been utilized by most of the largest biotherapeutic and pharmaceutical companies throughout the globe. Additionally, we provide the same level of services to mid-sized and virtual companies in need of high-quality data.
How does CovalX conduct Disulfide Bond Analysis?
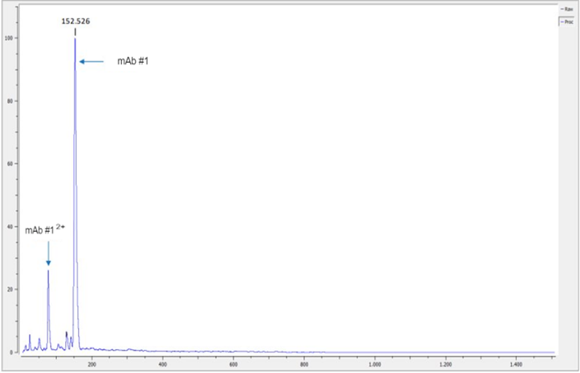
Intact Mass Analysis
As CovalX is known for our intact high mass measurements, we leverage this as an initial QC step when receiving large biotherapeutics for analysis. One of the first steps we conduct is an intact mass analysis to confirm that the molecule is correctly intact at the mass which agrees with the sequence provided. This is a rapid step that can confirm stability after transport and directly before disulfide mapping analysis.
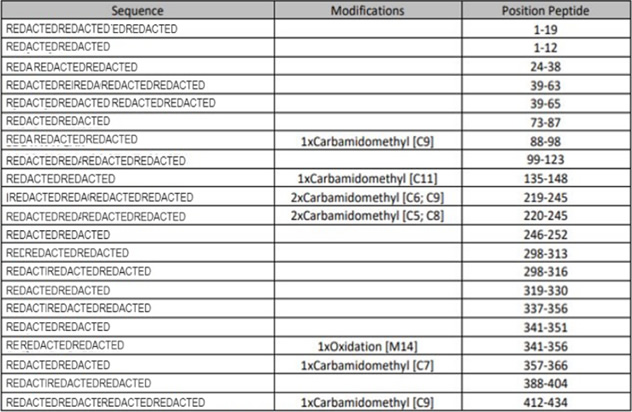
PMF – Peptides Detected and Modifications Seen
Using multiple enzyme digestion, nano-LC and high-resolution Orbitrap MS/MS analysis CovalX is able to accurately detect the peptides of the protein and identify several post-translational modifications upon these peptides. This data is included in a table format.
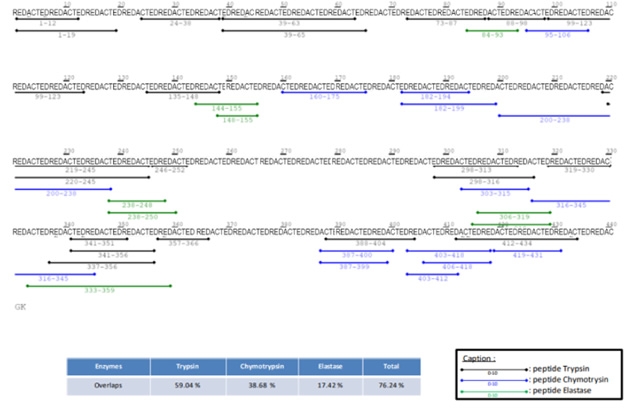
PMF – Sequence Coverage
From all of the peptides detected over the entire protein sequence these can be grouped and summarized in a custom-built chart to highlight the sequence coverage and any areas that may be undetected.
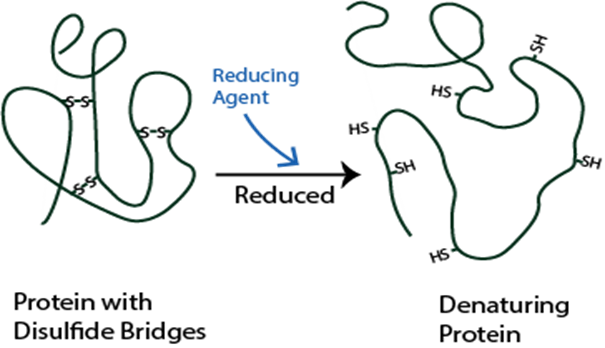
DTT Reduction – Mass change compared to non-reduced
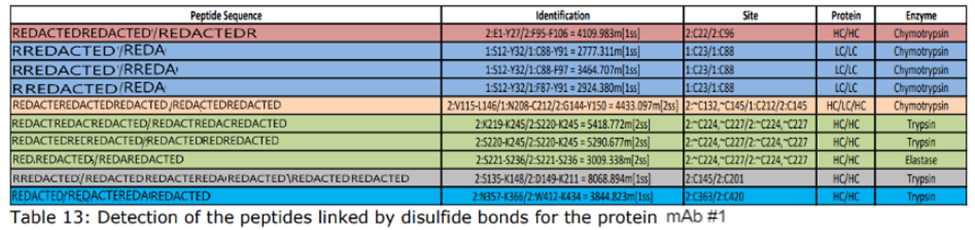
Table of Disulfides – ID specifically which residues are interacting
What is needed from the customer?
Sample types:
Recombinant proteins, purified proteins
Sequence information
The sequence of the protein to be mapped must be known. If it is not available from the client we can work to get this either from hybridomas sequencing or by DeNovo Sequencing.
Turnaround time
Three weeks or less from receipt of sample material
